Retinoblastoma

Definição
O retinoblastoma (RB) é um tumor da retina altamente maligno, cujo prognóstico depende do diagnóstico e tratamento precoces.
Causa
O aparecimento do tumor é devido à mutação no gene RB1. Para que o tumor se desenvolva é necessário que exista mutação nas duas cópias do gene. A primeira mutação pode envolver a linha germinativa ou estar confinada ao próprio tumor. Esta diferença determina duas formas diferentes da doença: RB hereditário e RB não hereditário. Estas duas formas de tumor têm características clínicas e epidemiológicas muito diferentes.
CLINICA GENÉTICA
Retinoblastoma hereditário
Apenas 10% dos doentes com RB têm história familiar da doença. Contudo uma mutação de novo na linha germinativa, contribui com mais cerca de 30% de casos. Por isso cerca de 40% dos doentes são portadores da forma hereditária do tumor.
Os doentes com a forma hereditária possuem uma mutação que envolve o gene RB1 em todas as células do organismo. Esta mutação é denominada da linha germinativa. Pode ser herdada de um progenitor, ou mais frequentemente, desenvolve-se numa célula no estádio embrionário do desenvolvimento do indivíduo afectado. A mutação da segunda cópia do gene RB1 acontece numa célula retiniana pruripotencial que vai dar origem ao tumor.
O gene RB1 tem um comportamento recessivo, ou seja, para que o fenotipo tumoral se expresse é necessário que ambos os alelos estejam afectados. Contudo ao nível individual, a mutação comporta-se de uma forma autossómica dominante, com uma penetração de 90%.
Os doentes com esta forma hereditária possuem um risco de 80 a 90% de desenvolver tumor ocular bilateral ou multifocal durante a infância; além disso possuem uma predisposição para, ao longo da vida desenvolver tumores não oculares. Cerca de 8% desenvolvem um segundo tumor intracraniano maligno primário, normalmente na glândula pineal – retinoblastoma trilateral. Cerca de 30% destes doentes acaba por desenvolver outro tumor não ocular até aos 40 anos. São mais frequente os osteossarcomas os fibrossarcomas e os melanomas.
Retinoblastoma não hereditário
Os restantes 60% dos doentes com RB são portadores de uma forma não hereditária ou somática da doença. Nestes doentes ambas as mutações do gene RB1 ocorreram dentro de uma única célula retiniana, resultando numa forma do tumor unilateral e unifocal. Nestes doentes não existe predisposição para o desenvolvimento de tumores não oculares.
Epidemiologia
É o tumor ocular mais frequente na criança. Ocorre em cerca de 1 para cada 20000 crianças e representa à volta de 12% de todos os tumores da infância.
Retinoblastoma não hereditário
Os restantes 60% dos doentes com RB são portadores de uma forma não hereditária ou somática da doença. Nestes doentes ambas as mutações do gene RB1 ocorreram dentro de uma única célula retiniana, resultando numa forma do tumor unilateral e unifocal. Nestes doentes não existe predisposição para o desenvolvimento de tumores não oculares.
Epidemiologia
É o tumor ocular mais frequente na criança. Ocorre em cerca de 1 para cada 20000 crianças e representa à volta de 12% de todos os tumores da infância.
A idade média de diagnóstico é 13 meses para as formas bilaterais e 24 meses para as formas unilaterais. Nos doentes diagnosticados em função do rastreio familiar a idade de diagnóstico é naturalmente mais precoce, sendo muitos casos diagnosticados perto do nascimento.
Cerca de dois terços dos tumores surgem antes dos dois anos e 95% surgem antes dos 5 anos. Depois dos 7 anos são muito raros.
Aspectos clínicos
Aspectos clínicos
É necessário distinguir a facilidade de diagnóstico entre famílias afectadas e famílias sem história da doença. Entre os filhos de pais afectados, o diagnostico é habitualmente mais precoce, numa fase em que o tumor é mais pequeno e oferece melhor oportunidade de tratamento para preservar o olho e a visão.
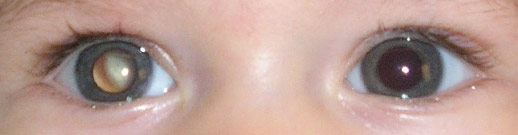
- Formas de apresentaçãoNas famílias sem historia de retinoblastoma, o tumor é diagnosticado acidentalmente ou após ter sido notada alguma alteração no olho afectado.O sinal de apresentação mais comum do tumor continua a ser a leucocória (pupila branca), em 60% dos casos. A segunda alteração mais frequente nas crianças afectadas é o aparecimento de um estrabismo. Outras formas de apresentação mais raras incluem: inflamação intraocular, neovascularização da íris, hifema (sangue na câmara anterior), celulite orbitária e glaucoma.Nos países desenvolvidos, a doença raramente se apresenta com evidência de extensão extraocular, doença metastática ou com sinais de envolvimento do sistema nervoso central.
- Formas de crescimento do tumorQuando não tratado, o tumor cresce de forma a preencher a cavidade ocular, e eventualmente a disseminar para fora do olho.O RB exibe 4 formas de crescimento. O padrão endofítico caracteriza-se por um crescimento para a cavidade vítrea, que facilita o diagnostico pela sua visualização directa. No padrão exofítico há um crescimento na direcção da coroide, elevando a retina, manifestando-se como um descolamento seroso da mesma. Na prática, a maioria dos tumores exibe características comuns aos dois padrões de crescimento – padrão misto. O quarto padrão de crescimento é uma forma difusa, rara (representa 1 a 2% dos casos), em que o tumor infiltra a retina produzindo um espessamento em placa; nesta forma, os sinais clínicos são muito subtis provocando muitas vezes um atraso do diagnóstico.As células do RB são pouco coesivas. Por esse motivo espalham-se com frequência para o vítreo (sementeira vítrea) ou para o espaço sub-retiniano, dependendo da direcção do crescimento do tumor. Podem ainda disseminar para longe do tumor primário, como locais distantes da própria retina (mimetizando um tumor multicêntrico), para a cápsula do cristalino ou mesmo para a câmara anterior (podendo complicar-se de glaucoma ou uveíte)
- Diagnóstico diferencialNos casos típicos o diagnóstico é fácil. Contudo existe um número significativo de casos em que o diagnóstico de retinoblastoma não se confirma. As doenças que mais vezes levam ao diagnóstico errado de retinoblastoma são a persistência de vascularização fetal (PVF), a Doença de Coats e a inflamação (granuloma) por toxocara.
- Avaliação OftalmológicaPerante a suspeita da presença de retinoblastoma o Oftalmologista deve fazer uma avaliação cuidada do potencial visual do olho ou olhos afectados. Este é um aspecto determinante para futuras decisões terapêuticas.O exame Oftalmológico deve ser o mais detalhado possível em função da idade e da colaboração da criança. Deve incluir a procura de sinais associados à presença do tumor.A observação do tumor deve ser efectuado sob dilatação pupilar com uma observação cuidada de todo o fundo ocular incluindo a periferia da retina. Sempre que possível deve ser obtido o registo fotográfico a cores do tumor, com uma câmara de contacto e com uma lente de grande abertura. Este registo é fundamental para a discussão das estratégias terapêuticas e para monitorizar a resposta às terapêuticas.
- Exames auxiliares de diagnósticoA ultrassonografia é um instrumento extremamente útil na avaliação do retinoblastoma. No modo A, é possível observar ecos de alta intensidade dentro do tumor, correspondentes às áreas de calcificação. O modo B da ecografia demonstra a presença de massa intraocular com ecos altamente reflectivos espalhados dentro do tumor e com atenuação do sinal atrás dele. Permite ainda diagnosticar a presença e a extensão do descolamento de retina associado.A tomografia computorizada e a ressonância magnética confirmam o diagnóstico e permitem avaliar a extensão extraocular do tumor. A tomografia é particularmente sensível na demonstração das calcificações intra tumorais características do retinoblastoma, na avaliação da sua extensão para fora do globo ocular, e na avaliação da órbita e das estruturas ósseas vizinhas.A ressonância magnética é particularmente importante na avaliação da extensão do tumor ao longo do nervo óptico e no sistema nervoso central.Na maioria dos casos a pesquisa exaustiva de metástases está reservada para doentes que apresentam características de doença muito avançada. É sempre obrigatório pedir a avaliação por um oncologista pediátrico.
Tratamento
O tratamento deve ser individualizado para cada doente. É determinado por vários factores que incluem o tamanho e a localização do tumor, o potencial visual do olho afectado, o envolvimento do nervo óptico, a expansão para a órbita e a presença de metástases.
O tratamento deve ser individualizado para cada doente. É determinado por vários factores que incluem o tamanho e a localização do tumor, o potencial visual do olho afectado, o envolvimento do nervo óptico, a expansão para a órbita e a presença de metástases.
Muito resumidamente poderemos dizer que na presença de tumores pequenos (tamanho inferior a 3 milímetros) se aplicam unicamente tratamentos locais com Laser ou crioterapia. Para os tumores maiores e sobretudo na presença de tumores múltiplos ou bilaterais é preferível efectuar quimioterapia para reduzir o tamanho do tumor (quimiorredução) e só depois se efectuam tratamentos locais com laser, crioterapia ou radioterapia. Nos tumores muito extensos, normalmente envolvendo mais de 50% do globo, ou em que existe opacificação dos meios ou neovascularização da íris faz-se a enucleação do olho afectado.
Com a grande evolução terapêutica das últimas décadas, podemos dizer que embora o objectivo primário do tratamento do retinoblastoma seja salvar a vida da criança afectada, na maioria dos casos o objectivo passa também por preservar o olho atingido e garantir a melhor função visual possível.
Vigilância após tratamento
A forma hereditária de retinoblastoma encerra um risco elevado de aparecimento de outros tumores da retina durante a infância. Daqui decorre a necessidade de uma vigilância apertada durante este período. Acresce que a forma familiar está associada ao aparecimento de outros tumores malignos ao longo da vida. A periodicidade dos exames de vigilância deve ser personalizada, deve envolver equipas multidisciplinares e deve manter-se ao longo da vida.
Prognóstico
O prognóstico é actualmente excelente no que diz respeito à sobrevivência, tendo também uma morbilidade ocular e visual muito diminuída. Nos países desenvolvidos a taxa de mortalidade caiu para valores inferiores a 10%.
O prognóstico a longo prazo é pior nas formas hereditárias devido à predisposição para desenvolver tumores exraoculares; é por isso necessária uma vigilância cuidadosa relativamente ao aparecimento desses tumores.
Na perspectiva da função visual houve uma melhoria muito significativa com o desenvolvimento de tratamentos que preservam a visão e evitam a enucleação.
Apesar da grande melhoria no prognóstico, o retinoblastoma metastático continua a ser de difícil tratamento. O factor mais importante para melhorar neste aspecto é a sua detecção precoce.
Na presença de envolvimento da glândula pineal, o prognóstico é bastante mau, com uma taxa de mortalidade aos 5 anos de praticamente 100%.
© ILUSTRAÇÕES: ORLANDO POMPEU
Desenvolvido por Sistemas do Futuro